

MDSPEC: Molecular Dynamics simulations of ion-matter interactions
Introduction
When neon ions strike a plastic surface, the resulting damage is anything but smooth and plays out at the scale of individual atoms. To capture those fleeting collisions, Authors; Pol Gherardi, Matthias Rupp and Jean-Nicolas Audinot, turned to MeluXina, Luxembourg’s national supercomputer, to support their large-scale molecular dynamics simulations. Their work follows Ne⁺ ions as they penetrate, displace and sputter atoms from polyethylene, revealing the full story behind surface damaged. The resulting insights will enable building better mass spectrometers for organic materials.
The Challenge of This Research
Recreating realistic ion–surface collisions in silico means overcoming three key hurdles:
1. System size and memory limits
A representative slice of polyethylene, measuring up to 20 nm by 20 nm by 15 nm, contains roughly 700 000 atoms. Tracking each carbon and hydrogen under high-energy impact pushes both memory and compute to their limits.
2. Reactive chemistry fidelity
As Ne⁺ ions collide, bonds snap and reform in femtoseconds. Capturing that chemistry accurately they employ a ReaxFF potential tuned with 549 parameters for C–H–O interactions.
3. Runtime versus resolution
To see damage evolve, simulations must run hundreds of thousands of timesteps without letting communication overheads or I/O slow things to a crawl.
The MeluXina Approach
To avoid relying on brute force, the team designed a simulation strategy that is both computationally efficient and scientifically accurate:
1. Spatial domain splitting
With the help of MeluXina’s massage-passing-interface (MPI), rather than a single massive block of polymer, the 700 000-atom system is divided into 16 smaller regions. That way the material is handled by 16 MPI tasks per node, ensuring efficient load distribution for parallel computation and avoiding bottlenecks.
2. High-fidelity force field
The finely tuned ReaxFF potential captures every collision cascade, captures every detail. From the initial ion collision to the ejection of atom clusters, every bond scission and cascade is accounted for. The results will be compared to observations from sputtering experiments.
3. Timed simulation batches
Simulations were conducted in batches of 10,000, 50,000, and 100,000 timesteps, across subsystems ranging from a 1,500-atom model to a 48,000-atom sample. This setup allowed for direct comparisons between system sizes and yielded useful performance data.
What They Learned
Running these simulations on MeluXina continues to deliver both scientific insights and practical benchmarking information:
1. Atomic Scale snapshots
The simulations provide frame-by-frame accounts of Ne⁺ ions striking the polymer, embedding themselves, displacing atoms, and distorting molecular chains. These sequences offer far more detail than any experimental tool can provide.
Picture Impact ion approaching the polyethylene surface (left) and ejected polymer fragments marked as sputtered material (right), revealing how Ne⁺ collisions dislodge atom clusters at the molecular scale.
Impact ion approaching the polyethylene surface (left) and ejected polymer fragments marked as sputtered material (right), revealing how Ne⁺ collisions dislodge atom clusters at the molecular scale.
2. Mechanistic Insights
By tracking individual atoms, the researchers map how energy propagated through the material. They identify penetration depths, recoil paths, secondary collisions, and ejection patterns. This helps to build a more complete explanation for experimental results.
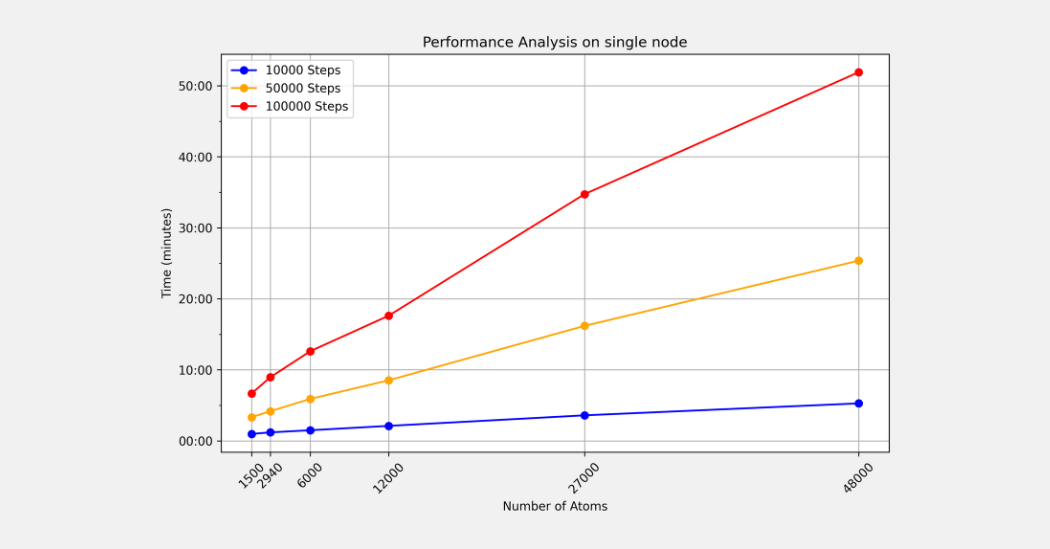
3. Performance benchmarks
Picture Measured runtimes on a single MeluXina node (128 CPUs) for 10 000 – 100 000 timesteps as system size grows from 1 500 to 48 000 atoms, illustrating strong scaling up to heavier workloads.
Measured runtimes on a single MeluXina node (128 CPUs) for 10 000 – 100 000 timesteps as system size grows from 1 500 to 48 000 atoms, illustrating strong scaling up to heavier workloads.
4. Roadmap for future studies
Thanks to these metrics and performance data, researchers can now tackle more ambitious simulations or investigate alternative chemistries with confidence in both scientific accuracy and computational feasibility.
Conclusion
This case study shows what becomes possible when precision science meets high-performance computing. MeluXina enabled the team to move beyond approximations and assumptions, uncovering the fine details of polymer damage caused by ion impacts. With accurate atom-scale trajectories and practical runtime data, the study sets a clear benchmark for future reactive molecular dynamics work. It provides not only a clearer understanding of how materials respond to extreme conditions, but also a tested and efficient method for treating them in silico.